This protocol describes the detailed experimental procedure for Double-Stem-loop LH-PCR. You may need to modify this protocol if you use different reagents or instruments for Double-Stem-loop LH-PCR.
1.Introduction
In Double-Stem-loop LH-PCR method, total RNA is extracted from the cells in which a given sncRNA is to be quantified (Fig 1). From this RNA mixture, the single-stranded sncRNA in question is captured by or hybridized to a special double-hairpin probe in which the single-stranded sequence complementary to the complete sncRNA in question is flanked by two hairpins. After sncRNA capture, one of the hairpins is cleaved by BamH I, leaving a five-base-pair stem that can dissociate at slightly elevated temperatures (50°C). The resulting single-stranded seven-base overhang is degraded by the single-strand-specific 3′-5′-exonuclease I (Exo I), yielding a nicked stem-loop in which the sequence hybridized to the sncRNA is protected from Exo I degradation only when the sncRNA is present. Thus, the sncRNA is changed into an equivalent DNA probe. In a subsequent PCR reaction, an oligo called “3′ terminus for PCR bridging” replaces the sncRNA from the capture oligo and is extended using the remaining part of the capture oligo as template. This newly synthesized stretch can then hybridize to the “5′ terminus for PCR bridging” oligo, resulting in PCR amplification of a longer fragment. Biotinylated primers that match the ends of this longer fragment are then used to label the long fragment with biotin on both ends. A second, post-PCR Exo I step then removes all remaining single-stranded oligos. The biotinylated PCR products are then transferred to a membrane and detected by avidin-alkaline phosphatase complexes (Li et al., 2014).
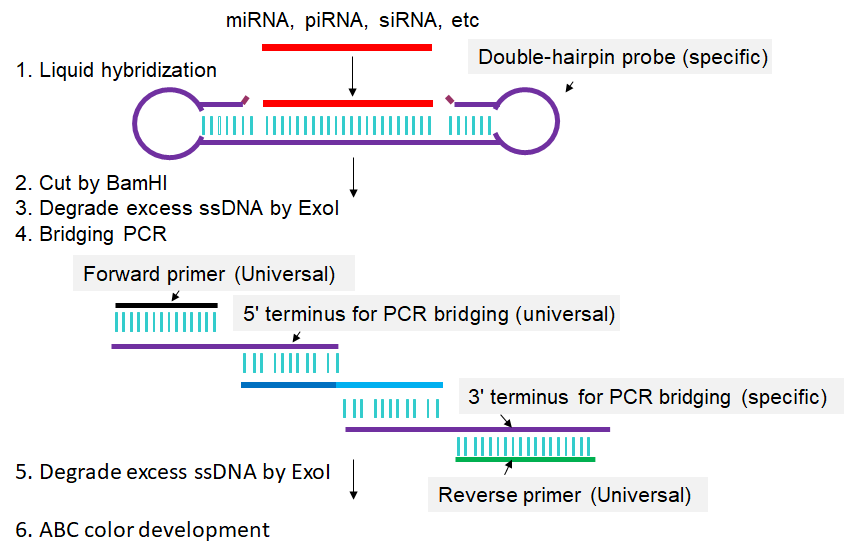
Fig 1. Scheme for Double-Stem-loop LH-PCR. Extracted RNAs are hybridized in buffer with a special double-hairpin probe. Next, BamH I is added into the mixture to cut the probes. Following that, exonuclease I (Exo I) is pipetted into the mixture to digest the nonhybridized probes. Then, bridging-PCR is performed to amplify the signals. Exo I is subsequently added into the PCR mixture to digest the excessive biotin-labeled primers. Finally, the mixture is spotted on a nylon membrane, incubated with alkaline phosphatase (AP)-labeled ABC (avidin–biotin complex), and developed with 5-bromo-4-chloro-3-indolyl-phosphate/nitro blue tetrazolium (BCIP/NBT) to produce visible colorimetric end products. For detailed experimental steps, please refer to the articles published by Li et al., 2014.
2. Material and Methods
2.1 Reagents and Equipments
(1) Oligonucleotide Primers. sncRNA specific primers can be designed or retrieved from sRNAPrimerDB (http://www.srnaprimerdb.com.). These primers are ordered from the company (miRNAPrimer: http://www.biootools.net/). All the primers are desalted and both UV absorbance and capillary electrophoresis are used to assess the quality of primer synthesis.
(2) Total RNA, or small RNA-enriched RNA, miRNA, siRNA, etc.
(3) Optical tube and cap strips.
(4) BamHI
(5) ExoI
(6) Standard ABC Peroxidase Staining Kit
2.2 Procedure
1. Liquid hybridization was performed as the LHCD method, that is, isolated RNAs were hybridized in hybridization buffer (1 X Exo I buffer) with double-hairpin probe (aslo named as two-hairpin DNA probes) at 42°C (Li et al. 2012). The hybridization mixture was further incubated with FastDigest BamH I (Fermentas) for 1 h at 37°C according to the manufacturer's instructions.
2. The mixture was digested with exonuclease I (NEB) for 5 min at 50°C and then for 1 h at 37°C. The short 50°C step must be performed to disrupt the short five-base-pair stem that remains on the capture oligo after Bam H I cleavage without disrupting hybridization of the miRNA to the capture oligo.
3. The digested mixture was used as the template for PCR, which was performed with KOD-Plus DNA polymerase according to the supplier's instructions. The PCR procedure was for 5 min at 94°C, then for 20 s at 94°C, for 20 s at 65°C, for 10 s at 68°C, X Cycles, and for 5 min at 68°C. The value of X was selected based on the requirement of the respective experiment.
4. The mixture of PCR amplicons was then subjected to digestion with exonuclease I for 0.5-1 h at 37°C.
5. The reaction mixture was spotted onto a nylon membrane and the LHCD method was applied (Li et al. 2012). Briefly, the membrane was incubated with ABC-AP, washed with TBST buffer, and developed with BCIP/NBP to produce colorimetric end products. For all tests, the spotted volume was 1 µL each.
2.3 Primers design
The used example sequences of RNA and DNA oligonucleotides (5’ to 3’) are listed as follows:
Experimental method H: Double-Stem-loop LH-PCR sRNA ID sRNA Sequence (5'-->3') Length (bp) GC (%) hsa-test-1 UGAGGUAGUAGGUUGUAUAGUU 22 36.36 PCR primer pairs Primer ID Sequence (5'-->3') Length (bp) GC (%) Tm (°C) Double-hairpin probe(specific) ACGTGCGAAAACGCGCGATAACTATACAACCTACTACCTCATTTATAGGATCCAATAAAAATTGGATCCTATAC 74 37.84 75.72 Forward primer(universal) TGGTACTGATGTGATGGACT 20 45.00 56.15 Reverse primer(universal) TCATATCACACAGCACCGAT 20 45.00 57.30 5'end for PCR bridging(universal) AGATGTGGTACTGATGTGATGGACTACGTGCGAAAACGCGCGAT 44 50.00 76.76 3'end for PCR bridging(specific) TCACCTCATATCACACAGCACCGATTGAGGTAGTAGGTTGTATAGTT 47 42.55 72.85 Total time used: 103.942933ms.
References:
1. Li X, Ni M, Zhang Y. Detecting miRNAs by liquid hybridization and color development. Methods, 2012, 85: 151-155
2. Li X, Ni M, Zhang C, Ma W, Zhang Y. A convenient system for highly specific and sensitive detection of miRNA expression. RNA. 2014, 20:252-9. doi: 10.1261/rna.040220.113.